Band Alignment, Electronic Structure of Materials Division
Head: Prof. Dr. Hongbin Zhang
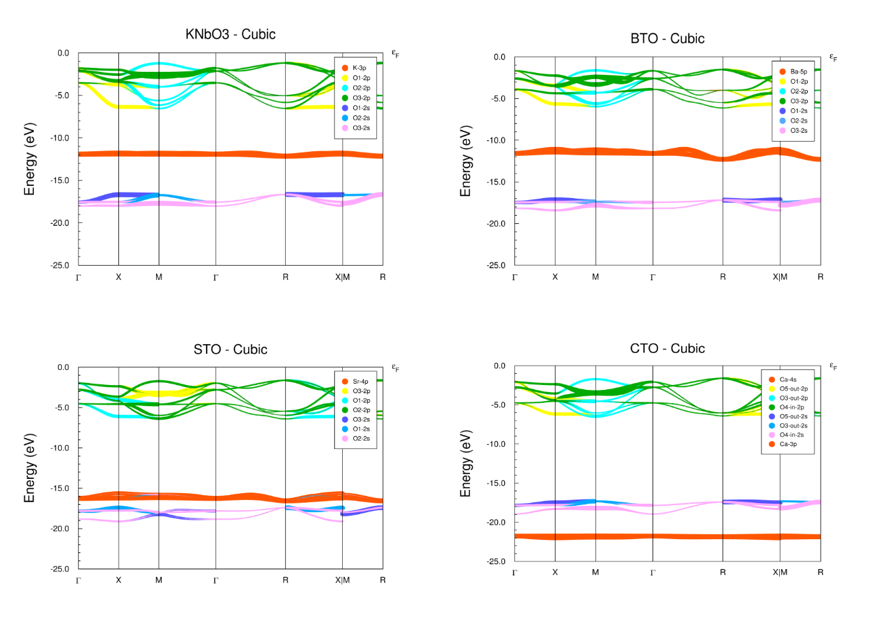
As we know, there is a unique relationship between physical properties of the system and the structure of the system, and similarly a unique relationship between band structure and physical characteristics, therefore it is completely reasonable to find a unique relationship between band structure and structural properties of the system. This is exactly our goal, namely to find a descriptor for the ferroelectrics and antiferroelectrics from the band structure. In terms of ferroelectrics, we studied strain, changing A_site element and Pseudo-John-Teller distortion as the probable candidates for the origin of distortions. One of the most important discoveries was emergence of A-site P-orbital between the O-2s and O-2p states in ferroelectrics, while this point does not exist in CTO as a quantum paraelectric. In order to gain a deeper understanding, we plan to study the PJTD for ferroelectrics to understand how valence and conduction bands hybridize during the distortions.
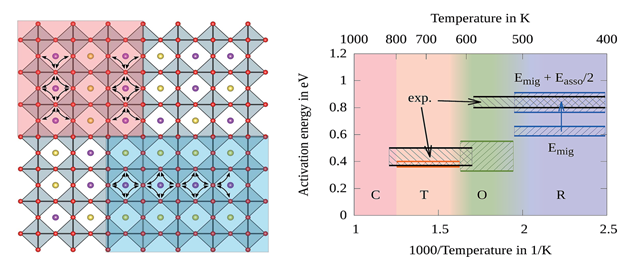
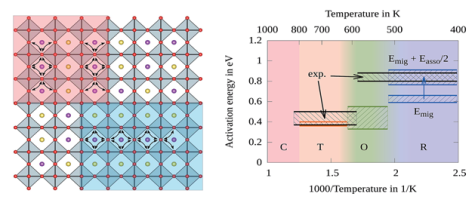
Usually in ferroelectrics, one or two distortion modes appear and usually they are coupled with each other in a simple way but in antiferroelectrics, there are more than two or three modes (for instance in AgNbO3 and NaNbO3 there are more than 9 modes and all of them are coupled with each other in a very complex way) , therefore studying ferroelectrics is not only good to figure out ferroelectric properties better, but also helpful in studying antiferroelectrics due to our previous information about how any of the distortion modes are coupled with the band structure.
In antiferroelectrics, due to large supercell, we need to study the unfolded band structure and we are going to study them in more detail.
Band Alignment, Electronic Structure of Materials Division
Head: Prof. Dr. Andreas Klein
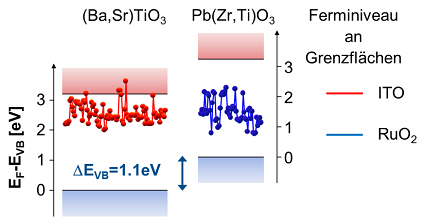
We investigate the relation between the position of energy bands and electronic defect states with antiferroelectric properties. These issues of the electronic structure are analyzed by means of photoelectron spectroscopy at interfaces and electrical conductivity measurements and are compared with theoretical predictions. The measurements are also important in order to understand the stability of high electric fields and the influence of doping and different electrode materials.
[1] R Schafranek et al. PbTiO3/SrTiO3 interface: Energy band alignment and its relation to the limits of Fermi level variation, Phys. Rev. B 84, 045317 (2011).
Defects, Materials Modelling Division
Head: Prof. Dr. Karsten Albe
This project investigates how the electronic and structural properties of antiferroelectric materials can be influenced or controlled by intrinsic and extrinsic point defects. Using first-principles calculations based on density functional theory (DFT), the formation energies of intrinsic and extrinsic point defects in different charge states are determined as a function of the Fermi energy and the chemical potential of the reservoir. The data can be used to determine electronic and thermodynamic defect levels and to predict defect equilibria. Furthermore, it is planned to investigate defect associates with dopants. In this context, the prediction of defect mobility plays an important role in material design, as the migration barriers of charged defects can be influenced by the location of the Fermi level. In addition to the chemical doping, which can affect the phase stability of ferroelectric and antiferroelectric phases, this work package will also investigate the role of polarons.